

About
Our lab aims at understanding how genetic changes between individuals can or cannot result in disease by quantifying the impact mutations have on protein aggregation and toxicity.
We are particularly interested in amino acid sequences that can adopt different conformations and undergo a process of self-assembly which results in distinct physical states.
Protein self-assembly
The aggregation of proteins into insoluble amyloid fibrils is a key process in the pathogenesis of a number of neurodegenerative conditions, such as Parkinson’s disease or Amyotrophic Lateral Sclerosis. However, examples of functional amyloid are also widespread in nature, especially across bacteria and fungi. Our work aims at systematically deciphering the sequence-dependencies of the process of aggregation in both functional and pathological contexts.
Proteins can also self-assemble into a more dynamic and reversible state through a process of condensation which is thought to contribute to the organization of the intracellular space. However, also for proteins that undergo liquid-demixing to form biomolecular condensates, the balance between function and dysfunction is far from clear. It is also unknown if and how condensates are precursors of insoluble amyloid-like states, and to which extent proteins are structured once in the liquid state.
Quantifying the impact of mutations at scale
In order to understand how mutations affect these delicate equilibria and to elucidate when and why a sequence starts aggregating or becomes toxic for the cell, our lab integrates experimental and computational approaches in different model systems. Recently, we have developed different Multiplexed Assays of Variant Effects (MAVEs) to quantify the toxicity and aggregation propensity of hundreds of thousands of protein sequences in vivo. By capturing the full landscape of the effects of mutations in a specific protein sequence we can guide clinicians to better diagnose and treat human disease, but we also reach a comprehensive mechanistic understanding of the process of amyloid formation and protein-induced toxicity. This translates in the possibility of rationally developing better targeted therapeutics, as well as in a set of fundamental principles that can guide protein design in bioengineering.
Developing novel strategies to report on protein conformation
In collaboration with the Lehner lab we also develop combinatorial mutagenesis approaches to study the interactions between mutations. We then use such genetic interactions to report on the conformation different proteins adopt as they start aggregating. Overall, we aim at generating exhaustive datasets that will give mechanistic insights on the process of protein aggregation, while also reporting on specific conformations and mechanisms leading to cellular toxicity. The massive amount of data we generate is used to train new models of protein aggregation. Our strategy is also amenable to tackle many intrinsically disordered proteins, which are particularly difficult to study in vitro. In this perspective, in vivo selection approaches such as the ones we develop can provide a unique opportunity to investigate these sequences in a systematic way.
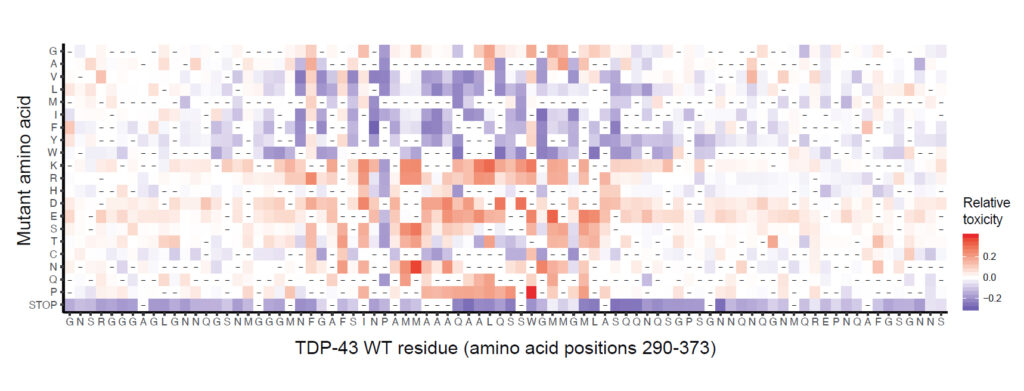
Map of the effect of mutations on toxicity of the TDP-43 Prion-like Domain.
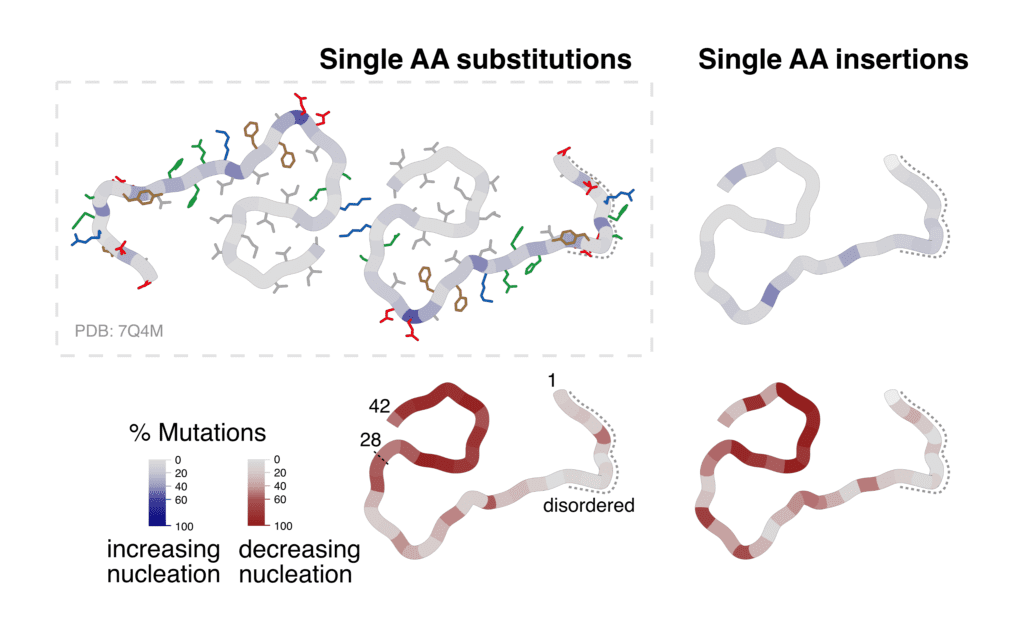
Percentage of substitutions and insertions increasing or decreasing amyloid formation of the Amyloid-Beta peptide, visualized on the cross-section of ex-vivo fibrils (7Q4M).
Staff
 ibecbarcelona.eu
ibecbarcelona.euProjects
| NATIONAL GRANTS | FINANCER | PI |
|---|---|---|
| AMYNDEL · Deciphering the consequences of different types of genetic variation in amyloid forming sequences by deep mutagenesis ( 2022-2025) | MICIU · Generación Conocimiento: Proyectos I+D | Benedetta Bolognesi |
| DeepAmyloids · Massively parallel mutagenesis to understand, predict and prevent amyloid nucleation in neurodegenerative diseases (2021-2024) | Obra Social La Caixa | Benedetta Bolognesi |
| FINISHED PROJECTS | FINANCER | PI |
|---|---|---|
| Poly-STOP · Developing modulators of protein aggregation in polyglutamine diseases by deep mutational scanning (2021-2022) | BIST · Barcelona Institute of Science and Technology | Benedetta Bolognesi |
| PRIOMUT · Escaneado exhaustivo de mutaciones en un dominio priónico para entender la toxicidad inducida por proteínas (2019-2021) | MICIU / Retos investigación: Proyectos I+D | Benedetta Bolognesi |
Publications
Check for more detailed information on the outputs of the Group at IBEC CRIS portal.
Publications list:
Equipment
- Thermo MaxQ 8000
Collaborations
- Priyanka Narayan
NIH-NIDDK - Xavier Salvatella
IRB, Barcelona - Fran Supek
IRB, Barcelona - Ben Lehner
CRG, Barcelona - Luke McAlary /Justin Yerbury
University of Wollongong, Australia
News

IBEC exhibe su innovación biomédica de vanguardia en el Health Revolution Congress 2026
El Instituto de Bioingeniería de Cataluña reunió a investigadores, inversores y líderes del sector con su evento “IBEC Health Revolution Day: Engineering the Future of Medicine” (Diseñando la Medicina del Futuro), como parte del Health Revolution Congress. A través de paneles de expertos, presentaciones tecnológicas y charlas científicas, el evento mostró tecnologías biomédicas de vanguardia y debatió cómo traducir descubrimientos científicos en soluciones sanitarias reales.

Un mapa mutacional sin precedentes revela cómo las mutaciones de la amilina influyen en la diabetes tipo 2
Un estudio del Instituto de Bioingeniería de Cataluña (IBEC) crea un atlas mutacional de la hormona amilina, cuya agregación puede dañar las células pancreáticas y desembocar en diabetes tipo 2. El equipo analizó 1.916 variantes de la proteína y comparó los resultados de este mapa mutacional con datos genéticos y clínicos de 500.000 personas. El trabajo, publicado en Nature Communications, abre nuevas vías para entender el papel de la genética en la diabetes y para diseñar fármacos más seguros y eficaces.

Tres proyectos del IBEC obtienen financiación ERC Proof of Concept para impulsar innovaciones en salud, neurociencia y tecnología biomédica
Dr. Benedetta Bolognesi, Dr. Irene Marco-Rius i Dr. Nicolò Accanto, investigadors principals de l’IBEC, han obtingut cadascun una prestigiosa beca ERC Proof of Concept. Es tracta d’un prestigiós finançament que concedeix el Consell Europeu de Recerca per explorar el potencial comercial i social de projectes de recerca duts a terme en institucions europees. Els tres projectes guardonats abasten des de noves plataformes per descobrir fàrmacs antiamiloides fins a tecnologies avançades d’anàlisi metabòlica i eines òptiques per estudiar el cervell en animals en moviment natural.

Bioingeniería para la medicina de precisión en el 18º Simposio del IBEC
El 18º Simposio anual del IBEC se centró en ‘Bioingeniería para la Medicina de Precisión’, una de las áreas clave de aplicación del IBEC. Fueron cerca de 300 las personas asistentes al evento, entre las que se encontraba personal investigador local e internacional. Un ambiente multidisciplinar en el que expertos y expertas de otros centros y la propia comunidad del IBEC tuvieron la oportunidad de presentar sus proyectos e intercambiar conocimiento.

El IBEC y el EMBL Barcelona coorganizan una jornada de colaboración para explorar sinergias
El Instituto de Bioingeniería de Cataluña (IBEC) y el Laboratorio Europeo de Biología Molecular (EMBL) han celebrado hoy una jornada de “matchmaking”. El evento ha reunido a investigadores e investigadoras destacadas de ambos centros con el fin de fomentar la creación de nuevas conexiones y promover el diálogo científico.

El IBEC y Antoni Muntadas presentes en la exposición «Animales invisibles» del Museo de Ciencias Naturales de Barcelona
El Instituto de Bioingeniería de Cataluña (IBEC) asistió a la inauguración de la exposición «Animales invisibles», una muestra que pone en valor la fauna extinguida, pero aún vívida y presente en el colectivo imaginario de la población. La exposición cuenta con una sección dedicada al tigre de Tasmania, una obra de Antoni Muntadas, en la que el IBEC ha colaborado de la mano de Benedetta Bolognesi, investigadora principal del instituto.

Siete laboratorios adicionales del IBEC alcanzan el nivel superior en la certificación My Green Lab
Siete grupos de investigación más del Instituto de Bioingeniería de Cataluña (IBEC) han sido certificados por My Green Lab, alcanzando la calificación más alta, el Green Level, por prácticas de laboratorio sostenibles. Con estas incorporaciones, el 70% de los laboratorios del Instituto ya están certificados.

El IBEC y el Hospital del Mar formalizan una nueva etapa de colaboración
Hoy ha tenido lugar la primera jornada de colaboración entre el Instituto de Bioingeniería de Cataluña (IBEC) y el Instituto de Investigación del Hospital del Mar, celebrada en el Parque de Investigación Biomédica de Barcelona (PRBB). El encuentro ha sido una oportunidad para compartir líneas de investigación, explorar áreas de trabajo conjuntas y consolidar una alianza estratégica con la firma de un convenio formal de colaboración entre el IBEC, el Hospital del Mar y el Instituto de Investigación del Hospital del Mar.

Crean un mapa del primer paso en la agregación de proteínas del alzhéimer que da pistas para futuras terapias
Se trata de un análisis a una escala sin precedentes: estudiaron más de 140.000 versiones del péptido Aβ42, que forma placas dañinas en el cerebro. Es el primer mapa que revela cómo las mutaciones afectan a una proteína en su estado de transición, una fase efímera y difícil de estudiar. El hallazgo abre nuevas vías para prevenir el alzhéimer y sugiere un método aplicable al estudio de otras proteínas implicadas en distintas patologías. El estudio, publicado en la revista científica Science Advances, ha sido una colaboración entre el Instituto Wellcome Sanger, de Reino Unido, el Instituto de Bioingeniería de Cataluña y el Centro de Regulación Genómica, en Barcelona.

IBEC coorganiza el simposio internacional de referencia en escaneo mutacional
Más de 300 personas asistieron la pasada semana a la octava edición del Mutational Scanning Symposium, celebrado 21 al 23 de mayo en el Parc de Recerca Biomèdica de Barcelona. Se trata de un evento internacional de referencia para debatir los últimos avances en tecnologías de escaneo mutacional y Ensayos multiplexados sobre el impacto de mutaciones . Este año, el simposio fue coorganizado por el Instituto de Bioingeniería de Cataluña, el Centro de Regulación Genómica y la iniciativa internacional Atlas of Variant Effects.
Jobs
Postdoctoral Researcher at the Phase Transitions in Health and Disease Research Group
PR_BB // Deadline: 24/08/2026
Post-doctoral Fellow at the Phase Transitions in Health and Disease Research Group
Ref: PR-BB // Deadline: 23/12/2025
Laboratory Techncian at the Phase Transitions in Health and Disease Research Group
Ref: LT-BB // Deadline: 9/12/2025
Senior Laboratory Technician at the Protein Phase Transitions in Health and Disease Research Group
Ref: SLT-BB // Deadline: 8/12/2025
Laboratory Technician in the Phase Transitions in health and disease Research Group
Ref: LT-BB// Deadline: 28/09/2025
Researcher in Training at the Phase Transitions in Health and Disease Research Group
Ref: RT-BB // Deadline: 30/07/2025
Research Assistant at Protein Phase Transitions in Health and Disease Research Group
Ref: RA-BB //Deadline : 16/01/2025
Research Assistant at the Protein Phase Transitions in Health and Disease Research Group
RA-BB // Deadline: 16/09/2024
Predoctoral researcher at the Phase Transitions in Health and Disease Research Group
Ref: PHDR_BB // Deadline: 12/08/2024
Post-doctoral researcher in the Protein Phase Transitions in Health and Disease Research Group
Ref: PR_BB // Deadline 30/10/2024