

About
Our lab aims at understanding how genetic changes between individuals can or cannot result in disease by quantifying the impact mutations have on protein aggregation and toxicity.
We are particularly interested in amino acid sequences that can adopt different conformations and undergo a process of self-assembly which results in distinct physical states.
Protein self-assembly
The aggregation of proteins into insoluble amyloid fibrils is a key process in the pathogenesis of a number of neurodegenerative conditions, such as Parkinson’s disease or Amyotrophic Lateral Sclerosis. However, examples of functional amyloid are also widespread in nature, especially across bacteria and fungi. Our work aims at systematically deciphering the sequence-dependencies of the process of aggregation in both functional and pathological contexts.
Proteins can also self-assemble into a more dynamic and reversible state through a process of condensation which is thought to contribute to the organization of the intracellular space. However, also for proteins that undergo liquid-demixing to form biomolecular condensates, the balance between function and dysfunction is far from clear. It is also unknown if and how condensates are precursors of insoluble amyloid-like states, and to which extent proteins are structured once in the liquid state.
Quantifying the impact of mutations at scale
In order to understand how mutations affect these delicate equilibria and to elucidate when and why a sequence starts aggregating or becomes toxic for the cell, our lab integrates experimental and computational approaches in different model systems. Recently, we have developed different Multiplexed Assays of Variant Effects (MAVEs) to quantify the toxicity and aggregation propensity of hundreds of thousands of protein sequences in vivo. By capturing the full landscape of the effects of mutations in a specific protein sequence we can guide clinicians to better diagnose and treat human disease, but we also reach a comprehensive mechanistic understanding of the process of amyloid formation and protein-induced toxicity. This translates in the possibility of rationally developing better targeted therapeutics, as well as in a set of fundamental principles that can guide protein design in bioengineering.
Developing novel strategies to report on protein conformation
In collaboration with the Lehner lab we also develop combinatorial mutagenesis approaches to study the interactions between mutations. We then use such genetic interactions to report on the conformation different proteins adopt as they start aggregating. Overall, we aim at generating exhaustive datasets that will give mechanistic insights on the process of protein aggregation, while also reporting on specific conformations and mechanisms leading to cellular toxicity. The massive amount of data we generate is used to train new models of protein aggregation. Our strategy is also amenable to tackle many intrinsically disordered proteins, which are particularly difficult to study in vitro. In this perspective, in vivo selection approaches such as the ones we develop can provide a unique opportunity to investigate these sequences in a systematic way.
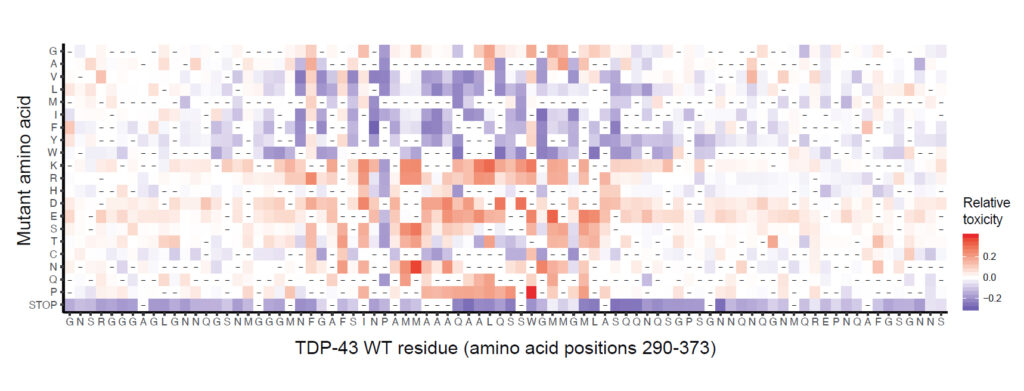
Map of the effect of mutations on toxicity of the TDP-43 Prion-like Domain.
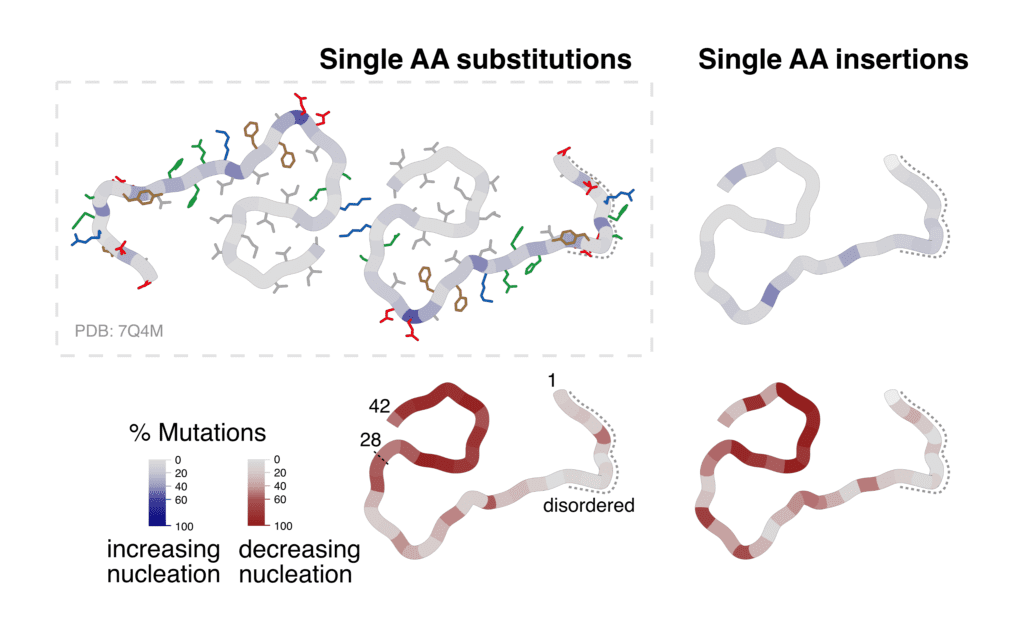
Percentage of substitutions and insertions increasing or decreasing amyloid formation of the Amyloid-Beta peptide, visualized on the cross-section of ex-vivo fibrils (7Q4M).
Staff
 ibecbarcelona.eu
ibecbarcelona.euProjects
| NATIONAL GRANTS | FINANCER | PI |
|---|---|---|
| AMYNDEL · Deciphering the consequences of different types of genetic variation in amyloid forming sequences by deep mutagenesis ( 2022-2025) | MICIU · Generación Conocimiento: Proyectos I+D | Benedetta Bolognesi |
| DeepAmyloids · Massively parallel mutagenesis to understand, predict and prevent amyloid nucleation in neurodegenerative diseases (2021-2024) | Obra Social La Caixa | Benedetta Bolognesi |
| FINISHED PROJECTS | FINANCER | PI |
|---|---|---|
| Poly-STOP · Developing modulators of protein aggregation in polyglutamine diseases by deep mutational scanning (2021-2022) | BIST · Barcelona Institute of Science and Technology | Benedetta Bolognesi |
| PRIOMUT · Escaneado exhaustivo de mutaciones en un dominio priónico para entender la toxicidad inducida por proteínas (2019-2021) | MICIU / Retos investigación: Proyectos I+D | Benedetta Bolognesi |
Publications
Check for more detailed information on the outputs of the Group at IBEC CRIS portal.
Publications list:
Equipment
- Thermo MaxQ 8000
Collaborations
- Priyanka Narayan
NIH-NIDDK - Xavier Salvatella
IRB, Barcelona - Fran Supek
IRB, Barcelona - Ben Lehner
CRG, Barcelona - Luke McAlary /Justin Yerbury
University of Wollongong, Australia
News

IBEC exhibeix la seva innovació biomèdica d’avantguarda al Health Revolution Congress 2026
L’Institut de Bioenginyeria de Catalunya va reunir investigadors, inversors i líders del sector amb el seu esdeveniment “IBEC Health Revolution Day: Engineering the Future of Medicine” (Dissenyant la Medicina del Futur), com a part del Health Revolution Congress. A través de panells d’experts, presentacions tecnològiques i xerrades científiques, l’esdeveniment va mostrar tecnologies biomèdiques d’avantguarda i va debatre com traduir descobriments científics en solucions sanitàries reals.

Un mapa mutacional sense precedents revela com les mutacions de l’amilina influeixen en la diabetis tipus 2
Un estudi de l’Institut de Bioenginyeria de Catalunya (IBEC) crea un atles mutacional de l’hormona amilina, l’agregació de la qual pot danyar les cèl·lules pancreàtiques i desembocar en diabetis tipus 2. L’equip va analitzar 1.916 variants de la proteïna i va comparar els resultats d’aquest mapa mutacional amb dades genètiques i clíniques de 500.000 persones. El treball, publicat a Nature Communications, obre noves vies per entendre el paper de la genètica en la diabetis i per dissenyar fàrmacs més segurs i eficaços.

Tres projectes de l’IBEC obtenen finançament ERC Proof of Concept per impulsar innovacions en salut, neurociència i tecnologia biomèdica
Dr. Benedetta Bolognesi, Dr. Irene Marco-Rius i Dr. Nicolò Accanto, investigadors principals de l’IBEC, han obtingut cadascun una prestigiosa beca ERC Proof of Concept. Es tracta d’un prestigiós finançament que concedeix el Consell Europeu de Recerca per explorar el potencial comercial i social de projectes de recerca duts a terme en institucions europees. Els tres projectes guardonats abasten des de noves plataformes per descobrir fàrmacs antiamiloides fins a tecnologies avançades d’anàlisi metabòlica i eines òptiques per estudiar el cervell en animals en moviment natural.

Bioenginyeria per a la medicina de precisió en el 18è Simposi de l’IBEC
El 18è Simposi anual de l’IBEC es va centrar en ‘Bioenginyeria per a la Medicina de Precisió’, una de les àrees clau d’aplicació de l’IBEC. Van ser prop de 300 les persones assistents a l’esdeveniment, entre les quals es trobava personal investigador local i internacional. Un ambient multidisciplinari en el qual experts i expertes d’altres centres i la mateixa comunitat de l’IBEC van tenir l’oportunitat de presentar els seus projectes i intercanviar coneixement.

L’IBEC i l’EMBL Barcelona coorganitzen una jornada de col·laboració per explorar sinergies
L’Institut de Bioenginyeria de Catalunya (IBEC) i el Laboratori Europeu de Biologia Molecular (EMBL) han celebrat avui una jornada de “matchmaking”. L’esdeveniment ha reunit investigadors i investigadores destacades d’ambdós centres per tal de fomentar la creació de noves connexions i promoure el diàleg científic.

L’IBEC i Antoni Muntadas presents a l’exposició “Animals invisibles” del Museu de Ciències Naturals de Barcelona
L’Institut de Bioenginyeria de Catalunya (IBEC) va assistir a la inauguració de l’exposició “Animals invisibles”, una mostra que posa en valor la fauna extingida, però encara vívida i present en el col·lectiu imaginari de la població. L’exposició compta amb una secció dedicada al tigre de Tasmània, una obra d’Antoni Muntadas, en la qual l’IBEC hi ha col·laborat de la mà de Benedetta Bolognesi, investigadora principal de l’institut.

Set laboratoris addicionals de l’IBEC aconsegueixen el màxim nivell en la certificació My Green Lab
Seven more research groups at the Institute for Bioengineering of Catalonia (IBEC) have been certified by My Green Lab, reaching the highest rating, the Green Level, for sustainable laboratory practices. With these additions, IBEC core facilities and 70% of the Institute’s laboratories are now certified.

L’IBEC i l’Hospital del Mar formalitzen una nova etapa de col·laboració
Avui ha tingut lloc la primera jornada de col·laboració entre l’Institut de Bioenginyeria de Catalunya (IBEC) i l’Institut de Recerca de l’Hospital del Mar, celebrada al Parc de Recerca Biomèdica de Barcelona (PRBB). La trobada ha estat una oportunitat per compartir línies de recerca, explorar àrees de treball conjuntes i consolidar una aliança estratègica amb la signatura d’un conveni formal de col·laboració entre l’IBEC, l’Hospital del Mar i l’Institut de Recerca de l’Hospital del Mar.

Creen un mapa del primer pas en l’agregació de proteïnes de l’alzheimer que dona pistes per a futures teràpies
Es tracta d’una anàlisi a una escala sense precedents: van estudiar més de 140.000 versions del pèptid Aβ42, que forma plaques nocives en el cervell. És el primer mapa que revela com les mutacions afecten una proteïna en el seu estat de transició, una fase efímera i difícil d’estudiar. La troballa obre noves vies per a prevenir l’alzheimer i suggereix un mètode aplicable a l’estudi d’altres proteïnes implicades en diferents patologies. L’estudi, publicat en la revista científica Science Advances, ha estat una col·laboració entre l’Institut Wellcome Sanger, del Regne Unit, l’Institut de Bioenginyeria de Catalunya i el Centre de Regulació Genòmica, a Barcelona.

IBEC coorganitza el simposi internacional de referència en escaneig mutacional
Més de 300 persones van assistir la passada setmana a la vuitena edició del Mutational Scanning Symposium, celebrat del 21 al 23 de maig al Parc de Recerca Biomèdica de Barcelona. Es tracta d’un esdeveniment internacional de referència per debatre els últims avenços en tecnologies d’escaneig mutacional i Assaigs multiplexats sobre l’impacte de mutacions. Enguany, el simposi va ser coorganitzat per l’Institut de Bioenginyeria de Catalunya, el Centre de Regulació Genòmica i la iniciativa internacional Atlas of Variant Effects.
Jobs
Postdoctoral Researcher at the Phase Transitions in Health and Disease Research Group
PR_BB // Deadline: 24/08/2026
Post-doctoral Fellow at the Phase Transitions in Health and Disease Research Group
Ref: PR-BB // Deadline: 23/12/2025
Laboratory Techncian at the Phase Transitions in Health and Disease Research Group
Ref: LT-BB // Deadline: 9/12/2025
Senior Laboratory Technician at the Protein Phase Transitions in Health and Disease Research Group
Ref: SLT-BB // Deadline: 8/12/2025
Laboratory Technician in the Phase Transitions in health and disease Research Group
Ref: LT-BB// Deadline: 28/09/2025
Researcher in Training at the Phase Transitions in Health and Disease Research Group
Ref: RT-BB // Deadline: 30/07/2025
Research Assistant at Protein Phase Transitions in Health and Disease Research Group
Ref: RA-BB //Deadline : 16/01/2025
Research Assistant at the Protein Phase Transitions in Health and Disease Research Group
RA-BB // Deadline: 16/09/2024
Predoctoral researcher at the Phase Transitions in Health and Disease Research Group
Ref: PHDR_BB // Deadline: 12/08/2024
Post-doctoral researcher in the Protein Phase Transitions in Health and Disease Research Group
Ref: PR_BB // Deadline 30/10/2024