


Myotonic dystrophy is a hereditary degenerative neuromuscular disease that occurs mainly in adults, affecting about 50,000 people in Spain. Symptoms range from difficulty walking to severe neurological problems, leading to progressive disability that unfortunately puts many of those affected in a wheelchair. This disease is very heterogeneous among patients (age of onset, progression, hereditary transmission, affected muscles), which makes the development of generic treatments especially complex.
Currently, drugs against myotonic dystrophy are developed in animal models such as Drosophila, the fruit fly, or in 2D cell cultures. They are later tested in mice and finally in patients. Unfortunately using these approaches, some of the drugs that have been developed have not been able to reach the last stages of clinical trials because they were not effective in patients, highlighting the difficulty of extrapolating results obtained with animals to humans.
Now, the team led by the ICREA Researcher Professor and head of the Biosensors for Bioengineering group at IBEC, Javier Ramón, and by his team’s postdoctoral researcher Juan M. Fernández, has developed for the first time a three-dimensional model for myotonic dystrophy, using bioengineering techniques and patient cells. The work, which is part of the doctoral thesis of the IBEC researcher Xiomara Fernández and has been carried out in collaboration with researchers from the INCLIVA health research institute in Valencia, has recently been published in the journal Biofabrication, and opens new avenues for finding personalized and effective treatments against this rare disease, and to date without cure. This work has been funded in part with funds from a fundraising campaign launched by IBEC from late 2017 to late 2019.
The new 3D model for myotonic dystrophy
 Researchers started from fibroblasts from patient’s skin, which are cells of the connective tissue responsible for maintaining its structure, that were previously “reprogrammed” to become myoblasts, muscle precursor cells. These myoblasts were placed inside a gelatine and cellulose matrix and the mixture was subjected to a kind of rubber stamp to give it a shape of elongated rectangles, simulating muscle fibers. Subsequently, the matrix was incubated under the ideal conditions for the myoblasts to continue their development.
Researchers started from fibroblasts from patient’s skin, which are cells of the connective tissue responsible for maintaining its structure, that were previously “reprogrammed” to become myoblasts, muscle precursor cells. These myoblasts were placed inside a gelatine and cellulose matrix and the mixture was subjected to a kind of rubber stamp to give it a shape of elongated rectangles, simulating muscle fibers. Subsequently, the matrix was incubated under the ideal conditions for the myoblasts to continue their development.
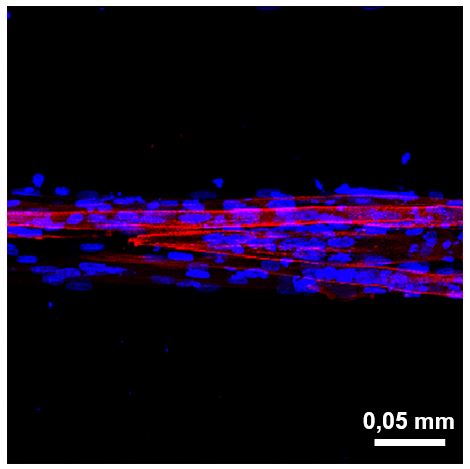
Image: In vitro confocal microscopy image of muscle developed from patient cells showing the three-dimensionality of the tissue. Cell nuclei are marked in blue and skeletal muscle in red.
What we observe is that, unlike classic cell cultures, the patient cells in our model are capable of fusing to form muscle fibers that adopt the 3D shape found in muscles and essential for their functioning.
Juan M. Fernandez
Ability to test drugs in a personalized and fast way
To evaluate the functionality of the new 3D model, the researchers have applied a drug, called AntagomiR-23b, to the muscle fibers. This drug has been previously developed by some authors of this work and in the not-too-distant future will enter the phase of clinical trials thanks to the Arthex Biotech company. They observed that both the molecular and structural characteristics of the fibers derived from the patient recovered their normal parameters when treated, validating their use for the development of new drugs.
In addition, this model has a low cost, its implementation is simple, and it can accelerate the process of developing new treatments, since it is faster compared to animal testing. This model represents a great step towards personalized medicine, since taking into account the enormous variability between patients is key when seeking treatments for this disease.
This new model will allow testing different drugs and administering to the patients the one that has given the best results with their own cells. In this way, the treatment will be personalized, more effective and with no need for animal testing.
Xiomara Fernandez
Myotonic dystrophy
The disease is caused by a mutation in the DMPK gene (Dystrophia Myotonica Protein Kinase) that causes the “sequestration” of essential proteins for the proper functioning of the muscles, especially the “Muscleblind” proteins. As its availability is reduced, it cannot normally perform its functions, including making the transition of other proteins from a fetal or chilhood phase to an adult phase. The consequence is that patients have proteins that do not function like those of an adult. Two examples are the insulin receptor, which in myotonic dystrophy patients does not work properly and causes diabetes, and the protein that regulates the entry and exit of chlorine from muscle cells, known as chlorine channels. Its malfunction is the cause of myotonia, which is the symptom that defines the disease and that causes patients to contract the muscle but have great difficulty in relaxing it.
Reference article: Xiomara Fernández-Garibay, María A. Ortega, Estefanía Cerro-Herreros, Jordi Comelles, Elena Martínez, Rubén Artero, Juan M. Fernández-Costa and Javier Ramon. Bioengineered in vitro 3D model of myotonic dystrophy type 1 human skeletal muscle. Biofabrication (2021), 13 (3), 035035.



